
|
| Recently Published Structures | ||||
|

|

|
|

|
|
Telomere G-quadruplex with mesoporphyrin PDB:4G0F |
Sec13:Sec16 PDB:4L9O |
Vps33:Vps16 PDB:4KMO |
Sec12 PDB:4H5I |
RapI PDB:4I1A |
Structure determinations fall into two classes: those with homology to existing structures in PDB; those with novel structures or with low sequence identity to any known structure.
You can get a quick indication of how homologous your molecule is to an existing structure by using a BLAST protein search and selecting the "Protein Data Bank" as the sequence database. If you find extended matches with greater than 40% sequence identity, you have decent odds of solving your structure by the method of molecular replacement. A single good data set can suffice to determine the structure. Most structures uploaded to PDB are now solved by this method. The method also gives a reasonable starting model to iteratively rebuild and refine the model toward the one respresenting your structure. A recent example of a structure determined by molecular replacement using the facility's resources is the Vps33:Vps16 subcomplex from HOPS. We prefer the programs Phaser and Molrep for most of our work but have also used Phenix.Rosetta for a recent difficult structure determination of the Rap phosphatase RapI - an external collaboration with Prof. Matthew Neiditch at Rutgers.
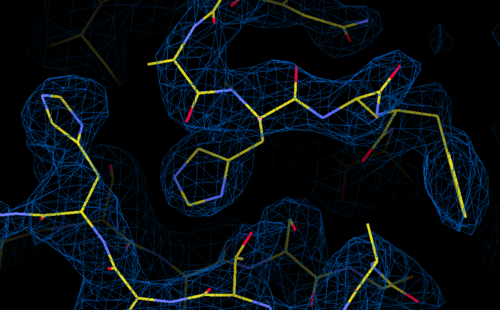
Sec12 experimentally-phased electron density map
from SelenoMethionine MAD data collected at the synchrotron
Software
Modern computers have got to the point where it's no longer necessary to drop $75,000 on a Silicon Graphics workstation to solve crystal structures. You can do structures on your MacBook, although not on your iPad. Silicon Graphics no longer even exists as a company. We can help you install and configure your own crystallographic software suite if you wish, if you choose to go this route rather than use one of the facility computers.
Linux and Mac OSX are the easiest options because they are both based on Unix operating systems which match the development environments for most of the useful software packages. If you're running Windows consider using a virtual machine to run Linux as your main crystallographic environment. We highly recommend CCP4, a comprehensive and mature software suite developed by academics for x-ray crystallography. CCP4 incorporates the Coot project, which is the graphical program we strongly recommend to manipulate your structure with. We also recommend Phenix which is an ever-expanding set of programs that while buggier than CCP4 often has more bleeding-edge methods (for better or for worse).
A [CCP4 + Coot + Phenix] installation should give you a fairly full-featured range of programs to solve and build structures with.