|
Macromolecular Crystallography Facility: Home |
|
Using HKL3000R for Data Collection at Princeton University
HKL3000R is Rigaku's version of the machine-driving variant of
HKL2000. We are currently running v714n - this is probably the last
version update that we will receive.
Historical Note and Caveat Emptor
The original version of this page dates to 2012. Subsequently I've edited
it in 2017 with the new machine and incorporated some material based on
Rigaku's vendor training of August 2017.
We first started using HKL3000 to drive the data collection part of
the system after an upgrade to the RAXIS-IV++ controller in 2012. The
initial quality of that software was relatively poor with several
functional bugs including one or two dangerous ones. The very first
version of this document was an extended rant on the theme of how it
was unwise that the vendor ship me that pile of junk, and how equally
unwise it was of the author (author name still redacted) to
regard it as functional.
Nevertheless HKL3000R did improve to the point where it was functional
to collect data on the old single axis RuH3R/RAXIS-IV++, or at least
if you didn't put too much faith in the strategy program(s). The penultimate version of the
RuH3R/RAXIS HKL3000R document is preserved for
posterity. This version refers only to the newer version that drives
the 007HF/AFC11/Pilatus 300K system. Parts of the original rant may
slip through the edit, or there may be new reasons to rant.
Hardware Note
The Rigaku RuH3R was a large-spot X-ray system with a 300µ beam,
an single axis goniostat and the relatively large but slow RAXIS IV++
detector. This stalwart system was 17 years old when we replaced it
and starting to show its age. The new system is 50x "brighter" than
the old one, where brighter is really a combination of microfocus generator
(007HF), better optics (Varimax HF) and a much, much better detector
(Dectris Pilatus3 R 300K) with 5-10x better DQE and a very fast
readout time. The detector is, however, smaller and this makes the
new partial 4-axis goniostat (ω φ 2θ and partial
χ) critical to mapping the desired slices of reciprocal space onto the
detector surface - basically the problem that small molecule
crystallography faces all the time, but to lower resolution and with
larger unit cells.
More extensive notes on the hardware are at
Xray-Rigaku-007HF-Pilatus-300K.html.
Controlling the Machine
The control computer is dual boot - one side of it is a Centos Linux
operating system running HKL3000R to control the machine and the flip side is a
Windows 7 operating system running CrysAlisPro. CrysAlisPro
can run in Small Molecule (SM) or Protein (PX) modes but for
macromolecular data collections we use Linux/HKL3000R.
This document doesn't cover CrysAlisPro - look elsewhere
for that.
HKL3000R effectively controls the goniostat and detector, indexes
data, calculates strategy and processes the data. In both Windows and
Linux contexts the XGControl program controls the generator including
start/stop and power ramping. The name of the control computer is
mol-xray3. It does not have other user accounts other than those used
for data collection. It does not have the ability to do "remote" data
collection although remote
monitoring of the data is on my To Do list.
Comparison with HKL2000
You might have encountered HKL2000 at the synchrotron - in some cases
it's used as the default method to process data and can be useful
because it shows the predictions vs observed reflections in real time.
HKL3000R is very closely related to the data processing in HKL2000 -
in fact the data processing parts are essentially identical. However
in HKL3000R the Collect tab contains the means to drive a multi-axis
goniostat (see Collect>Align and Collect>Manual). The Collect>Collect
sub-tab drives the actual data collection - this will obviously differ
from the synchrotron software (e.g. CBASS) used for this.
What's Changed Compared to the RAXIS IV++ System
- There are more axes on the goniostat - use Collect>Align to move
the goniostat into Crystal Mount position via the button of the same name or
to Zero Goniostat a position with all axes at zero degrees. Use Collect>Manual
to move the goniostat to some other position manually.
- There's a video camera - see it in Collect>Align, but note that this
is not a "click to center" arrangement and you must still adjust the goniostat.
- Rigaku Strategy program - present in the previous version, using it is now essential
since there's more than one axis to consider.
- Smaller detector means that you're more likely to use the Multi-Run Data Collection in the
Collect Tab (press the Multi button). For single runs you might need to specify the
φ and χ values in the fields on the Collect>Collect tab.
Other Manuals
- HKL2000 manual from HKL Research
- HKL3000 manual from HKL Research
as a PDF - doesn't deal with machine control and data processing in any significant way.
- Daniel Gewirth's older Denzo/Scalepack manual is sometimes more
useful in that it discusses more of the nuts and bolts of these two
programs, which are still the ones doing data processing in HKL2000
and HKL3000, but this older manual is now a decade out of date so some
of the recent updates in scaling are not reflected in it.
- HKL2000 manual from SSRL.
- HKL2000 at SER-CAT (APS).
TL;DR - a quick outline of workflow
- Initiate communication with device using Connect button in Collect tab
- Goniostat in mount position (Collect>Align); mount crystal and center
- Test shots at 0° and +89° for 10 or 30 seconds per 0.25°
- If OK then index using Index tab, select primitive triclinic in Bravais Lattice
- Refine as Fit Basic and Fit All then click Bravais Lattice to select likely correct lattice (symmetry)
- Refine with Fit All and then mosaicity
- In Strategy tab first select space group (if known) then launch Rigaku Strategy, edit to taste, exit strategy and Load DC to put strategy in Collect tab
- Start data collection via Collect Tab
- Check integration parameters (spot size, profile fitting radius etc), start integrating data runs
- Scale data to check for quality and potential point groups
- Finalize scaling (Error Scale Factor to change χ2), educated guess on space group
- Close program, go have celebratory beer
Using the Graphical User Interface (HKL3000R)
|
The computer is always left running, usually logged in, usually
running the Linux side of the dual-boot setup. Upon login, the
default Desktop has icons for the software on the left-hand side.
There are two gaudy diamond icons on the left labeled "HKL3000R" and
"HKL-3000R P300K Chi". You want to use the
HKL-3000R P300K Chi to control the machine. The
HKL3000R version of the program can be used to analyse data
you've already collected and cannot control the machine.
Other icons of interest are XGControl for controlling the
generator, ImageViewer which is apparently Rigaku's image viewing
program, and a standalone version of the Rigaku Strategy program -
probably not a lot of use without importing Denzo's idea of crystal
and goniostat parameters.
On the top of the window is the icon bar which you can use to open a
Terminal if you want to use standard Unix commands (e.g. for looking
in your home directory or managing data). That's the icon right next
to the System menu. The web browser (Firefox) is the icon next to
that. Since this is a data collection/processing machine, do not
install any additional software on this system without permission.
CCP4, SHELX, HKL2MAP, XDS are already installed if you want to do
other calculations. Other programs (e.g. Phenix) are likely to be
added in the near future.
|
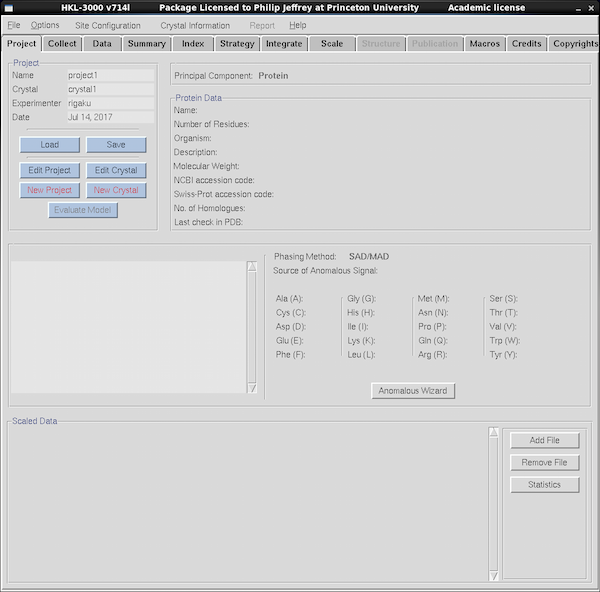
|
Quit any existing instances of HKL3000R to avoid accidentally
overwrite other people's data. Start the software by clicking on the
HKL-3000R P300K Chi icon. Underneath the top menu bar is a
series of tabs labeled Project, Collect, Data, Summary, Index, etc.
Click on each tab to reach the function for each. If you're screening
crystals you'll just use
Project and Collect. If you're collecting data or want
to auto-index your data you will use more of the tabs further to the
right. If you've used HKL2000 then the general layout is familiar.
HKL2000 is a data processing program that uses the underlying
programs Denzo and Scalepack, and HKL3000R is an extension of that program
to control the machine as well as process the data.
Use the Project and Crystal framework to keep images for
each protein under a distinct project name, with different crystals
being a new crystal within a project. Avoid making names absurdly long.
Click on the
Project Tab to manage projects and samples (shown at left).
You can create new projects, save existing ones, load old ones. When
HKL3000 starts it defaults to a generic project name - it doesn't save
your old project info so you should remember to save it manually
before quitting and load it after starting.
It's possible to
collect data and process it without changing the default project name.
Just make sure you specify a separate and unique data directory name
for your data and processing directories. I generally recommend
against doing this if you're actually collecting data since it
makes more sense to cluster related datasets into one project. Also,
NEVER modify someone else's project in this way.
To reload an existing project, use the Load button within the Project
tab and locate the relevant .sav file in the /data directory (scroll
past the subdirectory names to find them).
An alternative to this interface is to use the Project, Crystal and
Base Directory fields in the Collect tab. In all cases the data filenames
are
/data/Project_Crystal/Project_Crystal_0000.img
where Project and Crystal are taken from the values that you've given. The
"0000" gets automatically incremented, starting at 1, when you collect images. You
can use the Data Directory field in the Collect tab to provide a different directory
structure (e.g. grouped by name, lab etc).
|
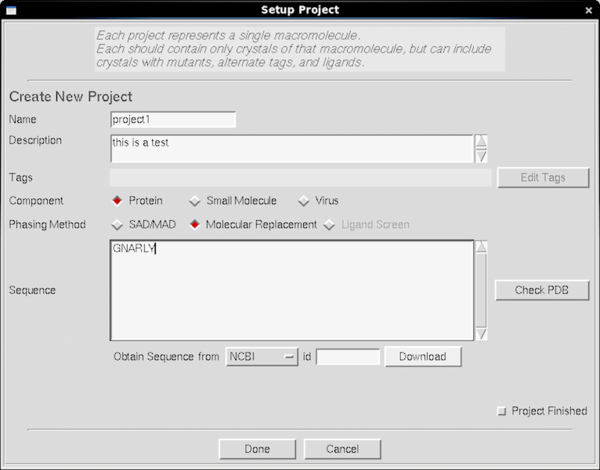
|
Here's what you see if you click on the New Project button.
Because HKL3000 has tries hard to be an overall framework for
structure solution, it can be more pedantic about projects than the
old CrystalClear software - however you don't need to give it the
sequence for project/crystal and can leave it blank. The main reason
to enter it is if you plan on using HKL3000R to solve your
structurefor you (not covered here) where the sequence is useful for
finding homologs or calculating the expected number of sulphurs. You
can solve Hen Egg-white Lysozyme using S-SAD from good tetragonal HEWL
crystals, but your crystal probably doesn't diffract like chicken
Lysozyme and when I try and do that party trick I do it on my own
desktop iMac using HKL2MAP or CCP4i - both of those are available on
mol-xray3 too. Most of the options on the project screen are a waste
of time for the simple purpose of crystal screening.
Nevertheless it's a good idea to use New Project and New
Crystal to manage subdirectories and filenames - or the equivalent
fields in the Collect tab - since the facility manager is very likely
to delete generic "project1" files if the disk starts to fill up.
|
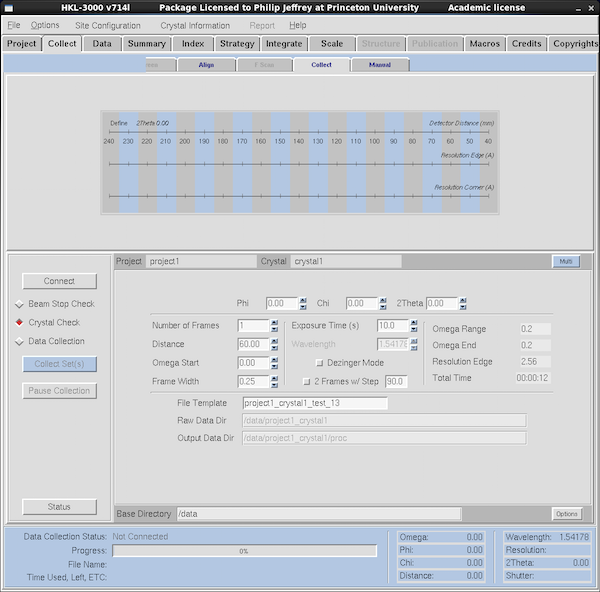
|
The Collect is by far the most important tab. To start doing
something productive you should mount your crystal and click on the
Collect Tab which brings up what you see at left. The buttons
on the left are the important ones. You need to press the Connect
button to get the program to talk to the machine - this
establishes connections to the servers in the generator itself. It
also initializes the detector and all axes on the goniostat when it
does so - this takes a while since the goniostat isn't all that fast.
LOCK DOWN THE PHI (φ) AXIS BEFORE CLICKING ON CONNECT.
Below the Connect button the radio buttons select what type of experiment you're doing.
Crystal Check loads default parameters for the sort of images
we shoot to test crystals. Data Collection are the sort of
parameters you'd use if you actually want to collect an entire
dataset. We'll come back to those settings, below. Make sure you define a Project
and Crystal either via the fields in this tab or via the Project tab.
The screen shot at left is from the Collect sub-tab within the Collect tab (yes, that's
confusing) but there are two other tabs within Collect that are useful: Align and
Manual.
|
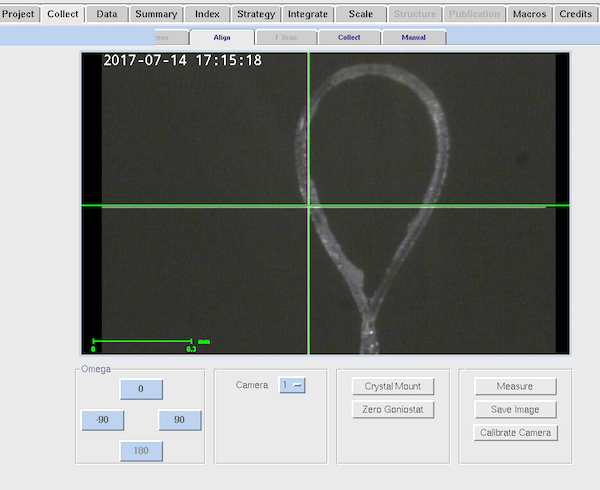
|
This is the Align tab within Collect.
Since the new system has a video camera for crystal centering you can
easily check on crystal status without getting anywhere near it during
exposure. You cannot adjust the centering from here. The most
useful part of the Align sub-tab is the Crystal Mount option that
drives the goniostat to the appropriate position to mount and remove
your crystal. You can also drive to ω=0, φ=0, χ=0,
2θ=0 and distance=100 using the "Zero Goniostat" button.
Loosen the grub screw on the phi axis to mount your crystal.
When centering your crystal be aware that for best results you need to
point a translation sled toward the camera and not toward you.
The camera is 30° away from the collimator axis (i.e. to
front left from the user point of view). You want to adjust the sled
that is perpendicular to the camera. Takes a little while to
adjust to if you're used to the former optical microscope being right
in front of you. If the crystal is moving weirdly on centering check
that you're doing this.
The goniostat has no vertical adjustment on the phi axis, but there is a certain
amount of vertical travel on the goniometer head. Be sure to mount your (empty) loop
before starting the experiment to make sure the height is approximately correct and your
pin length will work on this setup. The standard length of 21mm (sometimes called 18mm pin length)
will work just fine.
Remember to tighten the grub screw on the phi axis once your crystal
is centered.
|
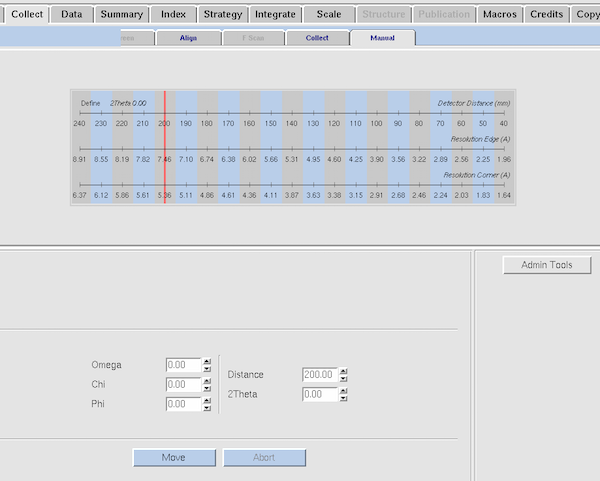
|
This is the Manual tab within Collect.
If you want to drive to a specific goniostat value, this is where
you'd enter it. Most of the time "Zero Goniostat" and "Crystal Mount"
within the Align tab would do the job. The current limits on
goniostat angles are: 2θ=+5 to -90°; φ= unrestricted;
χ= 0 to +45°. The ω axis limits are correlated with the
2θ position (potential collisions between the χ track and
the detector) - any angle where the χ axis forms an acute angle
with the 2θ track are excluded - but angles around
ω=zero° are always collision-free. Generally speaking be
ultra-cautious about driving the goniostat into positions manually -
drive them perhaps half-way and visually check for potential
collisions. The internal software checks should prevent
user-generated SNAFUs.
|
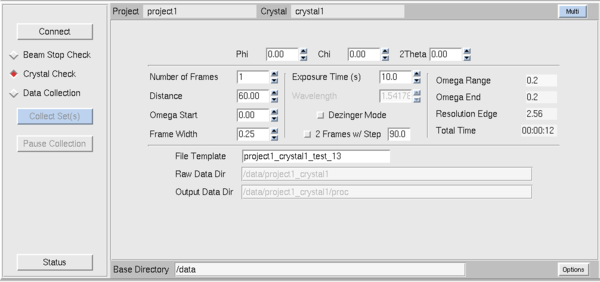
|
Now, back to the Collect sub-tab within Collect.
Below the Connect button the radio buttons select what type of experiment you're doing.
Crystal Check loads default parameters for the sort of images
we shoot to test crystals. Data Collection loads the sort of
parameters you'd use if you actually want to collect an entire
dataset. For Crystal Check you should set distance, number of frames,
omega (ω) start, frame width (0.25° perhaps - err on the
side of smaller images with the Pilatus, but not <<1/3 of the
mosaicity), exposure time (10 seconds is a good start) and click the
toggle if you want to shoot orthogonal pairs of images separated by 90
degrees. Limitations of the goniostat mean that sometimes it's better
to separate them by 80° or perhaps -90° since at
2&theta=0° an ω=+90° is in the forbidden range but
ω=89° or ω=80° is acceptable. Once you've changed
the values the blue button at left marked Collect Sets will be
enabled and you can collect the images. The new system is quite fast
- moving the goniostat is one of the slower steps but typical exposure
times are short (seconds) and readout is very fast (milliseconds).
With the old single axis goniostat on the RAXIS system, the rotation
axis was called phi (φ). Note that with the multi-circle
goniostat the scan axis is always omega (ω) rather than phi -
this is typical of multi-axis of goniostats where ω is the more
accurate axis used for scanning and the axes χ and φ are used
for repositioning only.
For actual data collection use the Data Collection button which
has many of the same parameters as Crystal Check. The current frame
is now displayed using the QT version of XDISP (it used to be
dtdisplay from Pflugrath's D*Trek) - where the contrast and image zoom
are the only significant features. This has a different GUI behavior
than HKL's X-windows based XDISP program and runs independently of it.
With the smaller detector and a more vesatile goniostat it's likely that
you'll be collecting data with multiple runs - in fact you'll probably
get better data if you collect with two different φ/χ
combinations rather than just one. I'll discuss strategy below - you
might have noticed the "Multi" button above-right on the panel and
that's where you define the multi-run data collection strategy.
Additional features on this tab are: "Status" button at the
lower left is the most useful - HKL3000 often rather mediocre
notification of what the machine is doing - I've seen it incorrectly
report what frame it's collecting. The Status button is sometimes a
more reliable source of information.
|
Most of the steps in data processing are analogous to my HKL Data Processing Guide which was written in 2002,
prior to the era in which y'all have been reduced to button clicking
for a living. The basic programs in HKL still underlie how HKL2000
and HKL3000 work - they just write scripts to run these programs via
the command line and parse the log files. Since I'm both a Luddite
and a Curmudgeon you'll frequently find me muttering while reading the
actual logs rather than the pretty little graphs.
Strategy Considerations

|
The Dectris Pilatus3 R 300K detector (P300K) is a smaller detector
(84x106mm2) than the RAXIS-IV++ (300x300mm2) and is
made of three modules stacked vertically. The image at left is from
the XDISP display program. The rotation axis on the home source is
vertical (i.e. ω). The smaller detector size in the instance of
high resolution diffraction may require you to collect multiple runs
of data to cover both low resolution and high resolution data, however
for many situations you could collect a medium-resolution data set
without offsetting the detector very much in 2θ.
This particular example is from definitive Hen Egg-white Lysozyme data
(tetragonal form) at 2θ=0° and a distance of 45mm. It's
usable to 1.9Å without offsetting 2θ and has exceptional
merging statistics. Tetragonal HEWL has a max cell dimension of
79Å and we could have collected a little closer.
Desired maximum resolution, point group symmetry and the largest
primitive cell dimension of your crystal all come into play with
strategy. The closest distance for a largest unit cell dimension
of A is A/2 with the closest distance between crystal and detector
being 34mm. This is better than the RAXIS-IV which was A/1.5. As
far as I know the neither the HKL3K nor Rigaku Strategy program
estimates the maximum resolution from the test images you have
collected - so you still have to eyeball it based on the images you
have taken (and/or data you have integrated and scaled). I recommend
adding 0.2Å to whatever it is you can see visually because the
detector has a habit of pleasant surprising you - and because you can
always reduce the resolution limit during processing, but you can't
incorporate data you didn't actually correct
Usually optimal data collection strategy is where the highest symmetry
axis of the crystal is almost parallel to the rotation axis. However
with the module boundaries being horizontal - perpendicular to the
rotation axis - this might give rise to systematic discs of missing
data - and this is likely to be more pronounced in low symmetry space
groups at lower resolutions. You might have to incline the symmetry
axis more, and this is where the strategy program comes in, and where
the partial 4-axis goniostat is of assistance. Collecting data at more
than one distinct crystal orientation (φ, χ) is likely going
to improve your actual data quality anyway.
|
Indexing and Generating a Strategy
You can process your data while it is being collected - recommended -
or you can process it later using the HKL3000R program icon (this version
doesn't control the hardware).
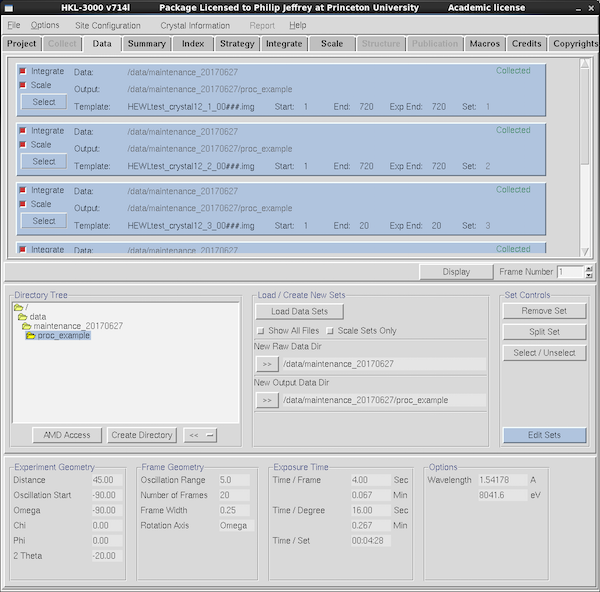
|
In the Data tab HKL3000 will load all data runs associated with
the project - both test images and data collection runs, and also for
data runs that are about to be collected. The blue boxes mark the
individual blocks of data. To remove irrelevant data runs, press
Select in the blue box and then Remove Set in the Set
Controls panel. The red check buttons within each blue data run
block offer control whether the data is scheduled to be integrated and
scaled. For initial indexing you need only one frame from one data
run so perhaps deselect the others - in particular the Project/Crystal
framework will often populate the Data window with data from all
samples for that project. Also when processing data after collecting
test oscillations, make sure you deselect those test frames since they
add nothing to the data.
If the data directory doesn't show up automatically, navigate to it
via the Directory Tree navigation panel at center-left, click to
select the directory and press the >> button next to New Raw
Data Dir. Select the processing subdirectory similarly and/or create
your own. Data collection parameters for the selected data set are
shown at the bottom of the tab.
To change the expected number of frames in a data set, select it using
the SELECT button, and click the EDIT SETS button (center-right).
Normally the correct number is found automatically, but you can use
this to exclude ranges of frames at the start or end of a set from
processing.
|
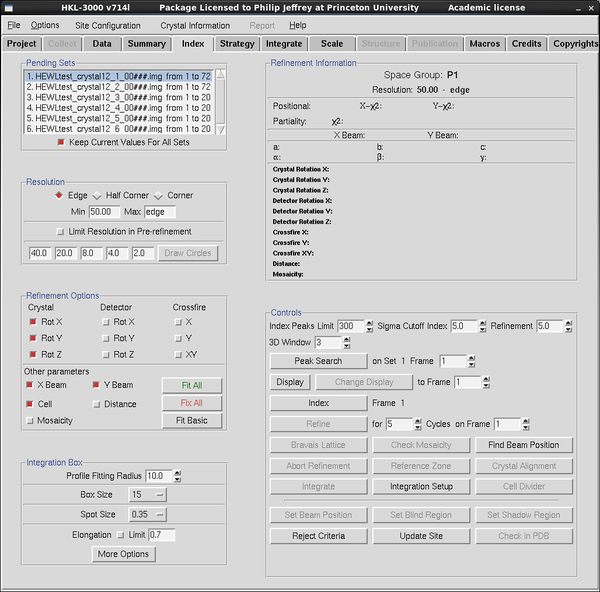
|
Indexing your diffraction pattern is a necessary precursor to strategy
calculation - symmetry axis orientations need to be known. Go to the
Index tab - verify that the data frame(s) listed are the ones
you want to index via the panel at top left - adjust via the Data Tab.
The text panel at top-right will contain the information for
indexed/refined unit cell parameters etc. The buttons below-right
control the workflow within Index. The check boxes below-left control
what parameters you want to refine to improve predicted and observed
spot positions, and a few things about spot size.
Click the DISPLAY button to view any frame within the run selected at
top left. Click PEAK SEARCH to pick spots on the frame. Unlike XDS
and MOSFLM, Denzo auto-indexes from a single frame. Most of the time
this works, but potentially less precise than extracting the information
from a pair (or range) or orthogonal images.
|
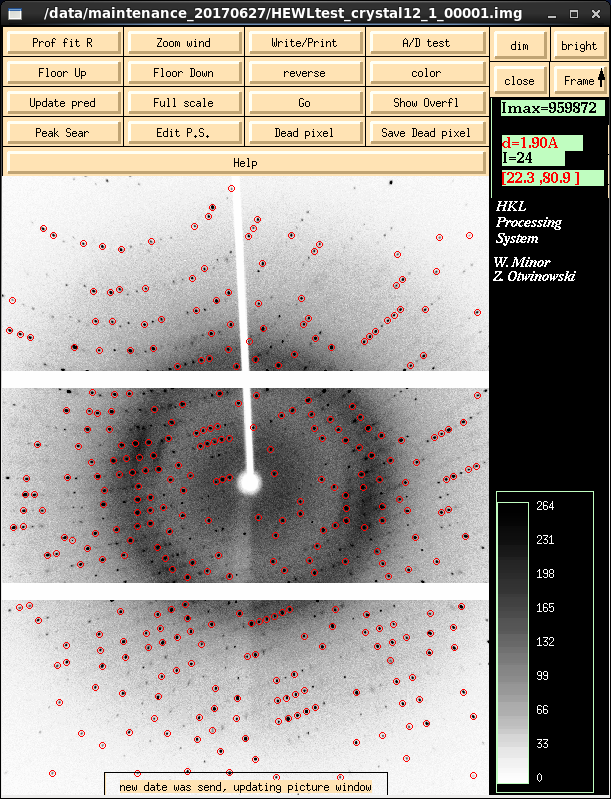
|
Denzo's graphical component - XDISP - is shown on the left in peak
search mode where it has searched for diffraction maxima based on
default criteria. These work well with strong diffractors. With
weaker diffractors and troublesome auto-indexing situations you want
to click on the Peak Sear button and pick more or less reflections
than the default.
You can also do Peak Search on images that are not the first one in
the data run, by entering the frame number in the box next to the peak
search button. For really problematic indexings I try every 10th
frame to get an indexing that I believe to be correct and work from
there. Denzo is smart enough to take into account the offset between
frame N and frame 1 when integrating the data.
It is possible to add spots from more than one contiguous set of images.
But you can't do it via the Peak Search button in HKL3000.
First, use DISPLAY to bring up the first image in XDISP. Press the
Peak Sear button in XDISP itself (lower left in button stack).
Then middle-mouse click on the Frame button at top right in
XDISP to load the next image, which will also peak search and
the peaks added to the list. Repeat for a few more frames. This way,
the peaks will be accumulated off multiple frames - and it also
appears to improve the mosaicity estimate during initial
auto-indexing.
Once you've got enough data spots selected - and sometimes I select an
abundance of spots on the frame and let Denzo sort out the wheat from
the chaff - press the INDEX button in the Index tab.
|
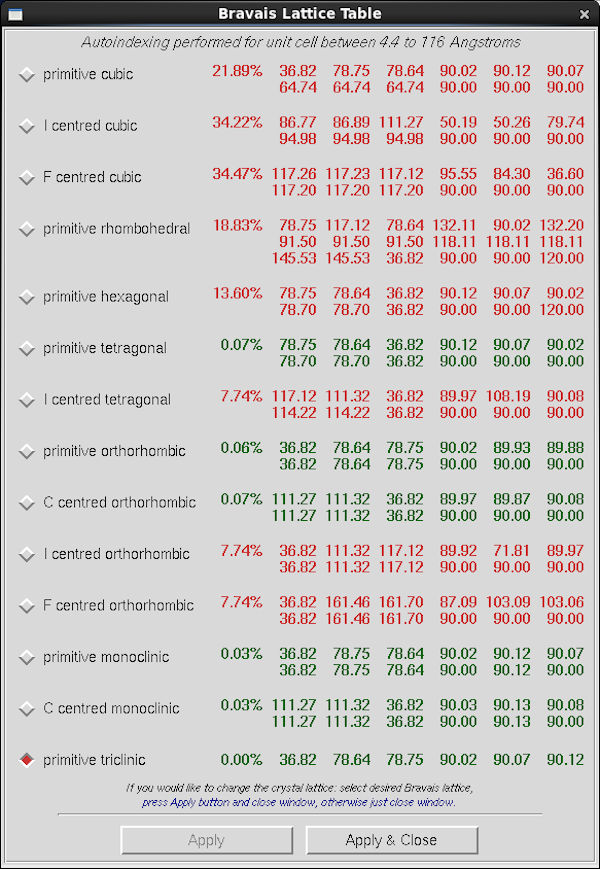
|
Denzo presents this table of possible indexings in the "Bravais
Lattice Table". The first one to pay attention to is primitive
triclinic all the way at the bottom - this is the inherent spot
arrangement on the frame and the largest value here corresponds to
your smallest inter-spot spacing. It's a Primitive (P) unit
cell which means that there aren't any of the systematic absences
present in C,F,I lattices that lead to a large proportion of the
reflections being systematically absent. On the lines above primitive
triclinic, Denzo permutes and potentially distorts that unit cell to
obey the symmetry-mandated requirements for each subsequent Bravais
lattice, with highest symmetry toward the top. The percentage value
it the degree of distortion required. Values in green required
relatively little distortion and those in red require more distortion
and are unlikely to be correct. Of the pairs of cell dimensions the
top one is the permuted cell and the bottom one is that cell after
it's "clamped" to the symmetry requirements. For primitive
tetragonal, which is what this unit cell is, the requirements are a=b
and α=β=γ=90°.
For weak auto-indexing results it's best to refine the auto-indexing
result in primitive triclinic first and then re-open this table using
the Bravais Lattice button in the Index tab. Success can be
influenced by the parameters for Index Peaks Limit and Sigma Cutoff
Index in the first line of the Controls panel within Index.
Be aware that the Bravais lattice table is responding only to the
location of diffraction spots in the frame and not their
intensity. While the highest symmetry choice is often the best one -
for HEWL the point group is 422 with a primitive lattice so primitive
tetragonal is indeed the correct choice - a minority of crystals show
pseudo-symmetry and the correct assignment of point group and unit
cell dimensions only becomes obvious during scaling. At which point
you must revisit the Bravais Lattice table and re-integrate the data.
To refine the initial indexing result (e.g. in Primitive Triclinic) you
select Fit Basic, refine a few times, then Fit All refine a
few times more and then revisit the Bravais Lattice table for your
best guess at the actual crystal symmetry/lattice system.
|
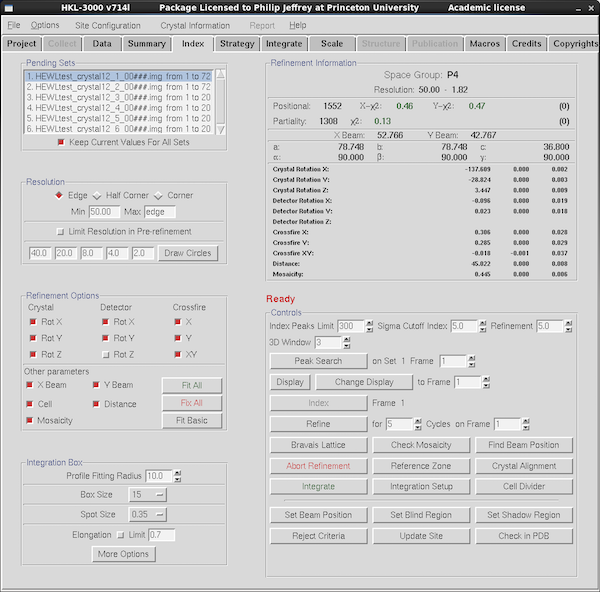
|
The current parameters are shown in the Refinement Information panel at top right.
For integration you basically only need to produce a good correlation
between predicted and observed diffraction maxima - some changes in
Bravais lattice can be accommodated in scaling even if you've
integrated it as the lowest symmetry primitive triclinic. However it
often stabilizes refinement to have the correct Bravais lattice
assigned, and it avoids you having to think about complicated
transformations (and applying them) in Scalepack later on.
To optimize the predicted vs observed maxima you need to refine the
initial auto-indexing parameters. The boxes toggled in the
Refinement Options panel at the left show what parameters are are
being refined the next time you press the Refine button. Some
eigenvalue-filtering hopes to reduce the correlations between
parameters but the idea here is to Fit Basic until convergence, add
in Distance and perhaps Fit All with mosaicity unchecked, followed
by Fit Basic with mosaicity checked, followed by Fit All again.
Mosaicity estimates are sometimes problematic for crystals with
streaky spots, and if the refined mosaicity climbs too high it will
likely cause problems with spot overlaps (spots whose centroids are
too close to each other on the frame). You can play games with not
refining mosaicity or you can redefine it at each integration step
using the Macros tab (not covered in this guide).
For weak data you'd probably do well to decrease the sigma cutoff for
data in refinement, as given in the Refinement box in the Control
panel to the right.
If you're unhappy with the spot predictions and wish to discard the
current parameters you'll need to press the Abort Refinement button
to go back and do peak searching or indexing again.
|
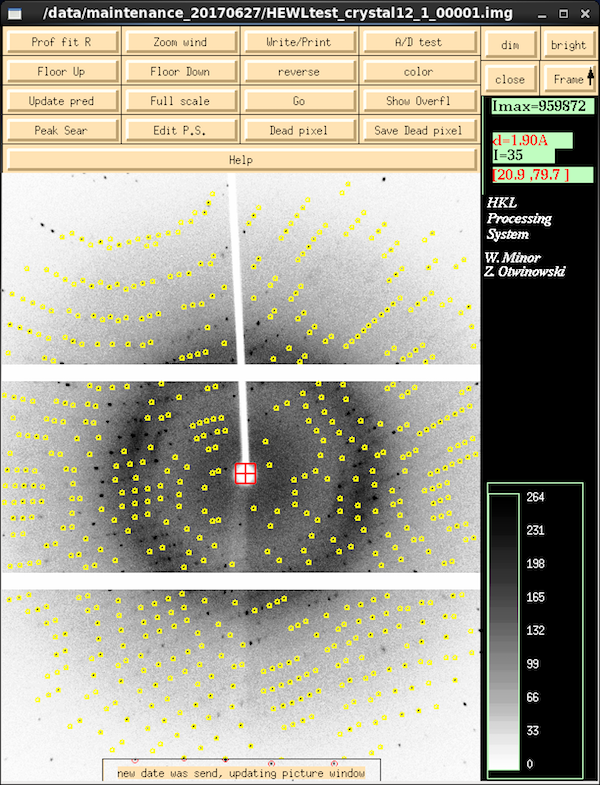
|
Once you've started refining parameters, XDISP changes to display the
predictions of spot centroids based on the refined parameter values -
toggle this using the Update Pred button to see if the predictions
(yellow, green, red) match the actual spot locations. Yellow centroid
markers are
partial reflections - reflections that are clipped at one or
both ends of their profile during the frame and whose diffraction
intensity is spread over multiple frames. Green reflections are
full reflections that complete their entire passage through
diffraction condition during the frame. On this image - collected
with 0.25° frame width on a crystal with 0.45° mosaicity -
every reflection is a partial. You only see a significant number of
fulls on thicker frames, particularly at higher resolutions.
Reflections with centroids that are red have issues - highly variable
background, overloads (unlikely with this detector), spots that are
too close to each other ("overlaps").
|
If the predicted spot centroids match the observed ones well enough you
can then move to the next tab - Strategy - to calculate the runs
necessary to collect a complete data set. Again, you'll have to use
existing images to assess the likely limit of diffraction based on your current
exposure time, although of course you can adjust this during the data processing step.
Strategy
You will be using Rigaku's strategy program rather than HKL3K's own
strategy program since the former is aware of the multi-axis
goniostat. Strategy makes use of the unit cell, space group and
crystal orientation that you've determined during auto-indexing. As
such, unless you really know what your space group is (for this HEWL
crystal it's P43212) then the strategy is really
an educated guess that must be verified during scaling.
|
|
|
After auto-indexing, click on the Strategy tab and
change the space group as necessary - inherently you're making a
decision on point group rather than actual space group at this stage.
By default Denzo will opt for the lowest-symmetry space group/point
group within the Bravais Lattice that you selected. In this case it's
space group P4 in point group 4 (Laue group 4/m). Tetragonal
lysozyme is actually space group P43212
in point group 422 (Laue 4/mmm) which affects the strategy since 422
is higher symmetry than 4. If you don't know what your space group
is, stick to the default. The worst that can happen is that you'll
collect more data than you need.
Click Rigaku Strategy to launch the program. Click Yes I
Want To Make Changes. In the sidebar are options for completeness
(recommend: 98% or greater), redundancy (recommend: 3 or greater),
resolution (you'll have to eye-ball that) etc. Click
Calculate Strategy to get the program to come up with something.
Data Processing
In a perfect world a data strategy program would guarantee that your
data collection will result in complete, redundant data without you
having to check. It's not a perfect world. As with synchrotron data
collections I strongly advocate checking the progress of your
data collection in real time and with the Pilatus system it's a lot
closer to real time than the RAXIS system. Crystals on the home source
don't damage anywhere near as fast as at a synchrotron (at least a few
days in the beam) but incomplete data is useless for most tasks and
there's no excuse for collecting it.
Denzo will pick up goniostat values from the frame header, so if
your crystal remains in the same position on the goniometer head
during the different runs you should be able to use the same relative
indexing - this is particularly important in point groups 3, 32, 4, 6
to avoid the relative indexing problem (see CCP4's reindexing page). This would show up in scaling
but understanding why it happens is very useful, particularly when
comparing multiple crystals.
Go back to the Data tab and select the runs you want to include
in data processing, likely excluding the test images you took when assessing
data quality or assessing a strategy. Index the data and refine the
parameters using the Index Tab in exactly the same way I outlined above.
|
|
Check again the correspondence between predicted and observed spot
centroids. Yellow and green reflection centroids are partials and
fulls, respectively. Red reflections are ones with a problem:
overloads (unlikely on the P300K); overlaps (spots that are too
close); spots with badly behaved backgrounds (too close to module
boundaries etc). If that looks OK be sure to check the spot
integration parameter on the lower left of the Index tab under the
Integration Box panel. Denzo, like other refinement programs, uses
learnt profile fitting to provide a better estimate of weak data by
estimating weak spot profiles by generating empirical spot profiles
from nearby strong spots. Profile Fitting Radius sometimes
needs to be increased for particularly weak or anisotropic data or
Denzo will refuse to integrate weak data with insufficent nearby
strong spots to estimate the profile from. Historically it also paid
attention to one of the sigma cutoffs (perhaps refinement?) shown in
the Controls panel - that defined the σ(I) boundary for what was
considered strong vs weak. Only strong spots are used to construct
spot profiles.
|

|
To get a look at the spot size, first click on Zoom Wind button in
XDISP to open the zoom window, then middle-mouse click somewhere on
the master image to center the view in the zoom window. Click on
Zoom In a few times to, well, zoom in. Click on Int Box to show
the integration box. The square is the background box that Denzo fits
a least-squares plane through to subtract the background from the spot
prior to integration. It's OK for those boxes to overlap - that's
controlled by the Box Size parameter in the Index tab. The box needs
to be several times the size of the spot to get a reliable background
estimate. The yellow circles are the spot centroid positions
(yellow=partial, same as before) and inside these are two concentric
circles around the spot, although it's not that obvious in this image
since the spots are only a few pixels wide. The central one is
controlled by the Spot Size parameter in the index tab. The point
spread function on the P300K is relatively small so spots are usually
quite small (and the pixels are larger at 170µm) but it's
important that the spot size encompass all of the reflection -
increase Spot Size as necessary after sampling various parts on the
image. If you change these parameters you have to do one cycle of
Refinement to update the view in XDISP. Caution: if you make the spot
sizes too large, adjacent spots will overlap each other and Denzo will
reject these spots as "overlaps". The spot centroids show up as red in this
case.
Are we done yet ?
If you've got the parameters refined so that the predicted spot
centroids match reality, you've selected the correct Bravais lattice,
and you've tweaked the spot size parameters as necessary you're pretty
much good to start integration. However if your crystal does
not diffract to the edge of the detector there's not much point
integrating fresh air - you can limit the resolution for integration
via the Resolution panel in the top left corner. Conversely if you
have really strong data (as this is) you could integrate into the
Half-Corner or Corner of the image, although you'd get far better data
coverage if you moved the detector closer (if possible) or moved the
detector further out in 2θ. You will need to do one cycle of
refinement, again, if you change the resolution limits.
|
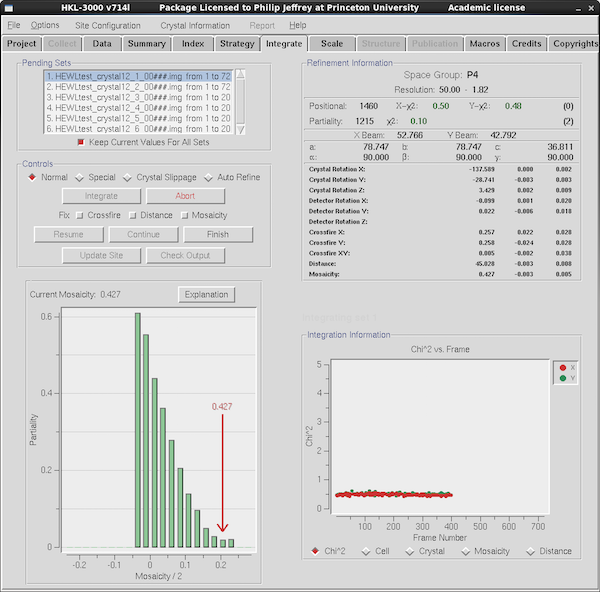
|
Go to the Integration Tab to start integration. Generally
speaking most of the parameters that affect integration are actually
on the Index tab. But if you particularly want to fix mosaicity
during integration (e.g. for smeared spots or crystals with other
types of large mosaicity issues) then this is the place to select it -
in the Controls panel at top left.
Check that the data runs listed in the table at top left are all the
ones you want to integrate, then press Integrate. If this is not the
first time you've integrated the data you'll be prompted if you want
to overwrite the ".x" files or create a new subdirectory. I nearly
always do the former. Denzo writes a file with extension ".x" that
contains the spots that are integrated on a particular image, as well
as the current integration and crystal parameters. Scalepack then
reads all these .x files to scale and merge the data.
During integration Denzo will happily wait for frames that are being
collected - just make sure it isn't hanging up at the end of data
collection waiting for frames that will never arrive. Denzo will
display the position residuals (chi2) for detector X and Y
between predicted and observed spot positions. Something less than 1.5
is desired here. The "Integration Information" panel at lower right
will show you positional chi2, unit cell parameters etc
during integration. It's not all that uncommon for one or more
parameters to drift during integration since the initial indexing was
done of one frame and the parameters are refined locally off
one or a very small number of frames - they aren't estimated over a
large angular range. Nevertheless you shouldn't see mosaicity or
distance wander very much (although mosaicity can be anisotropic). If
they do you might consider fixing them during integration.
Most of what goes on in the integration tab isn't all that exciting in
terms of monitoring data quality, but you still want to make sure
those residuals are small.
|
Scaling
Scaling your data is where you start to find out what your intrinsic
data quality is, so we need to define this: you want data that is
complete, that is of high precision, and has a reasonable
redundancy (aka multiplicity) so that the statistics are reliably
estimated and the scaling procedure is stable.
Inherently a number of semi-empirical parameters are refined to
minimize the intensity differences between observed reflections that
should have the same intensities based on symmetry. This requires an
expectation of what the error should be based on the strength
of the data and of what the likely point group symmetry is.
Symmetry: in this example we picked a Bravais lattice
(i.e. primitive tetragonal) which can accommodate two different point
groups, 4 and 422. The former has a single four-fold symmetry axis
and the latter also has two-fold symmetry axes perpendicular to the
four-fold. Alternatively speaking: point group 4 has eight
symmetry-equivalent reflections in a complete sphere of data and 422
has sixteen of them (assuming Friedel's Law holds - no anomalous
scattering). By picking a space group that is compatible with the
Bravais lattice you intrinsically assign a point group to the data:
space group P43 is in point group 4 and space group
P43212 is in point group 422.
Friedel's Law: Friedel's Law applies in the case where
anomalous scattering contributions are very small. It introduces a
center of symmetry in the data, so that reflections (h,k,l) and
(-h,-k,-l) have the same intensity. Elements C, H, N, O have
minimal anomalous scattering at CuKα wavelength (1.54Å)
but S, I, Lanthanides do not. For some well-diffracting proteins -
like the HEWL xtal here - you can actually use the very small sulphur
anomalous scattering signal to determine the structure but for weakly
diffracting proteins you can mostly ignore the presence of this signal.
Scaling: Scalepack is the program that does the scaling. There
are some behind-the-scenes analytical corrections for Lorentz and
polarization factors, air absorption etc but then Scalepack refines a
per-frame scale factor (k) and a per-frame B-factor (B) as a primary
method of reducing systematic differences in the data - i.e. the
differences in measured intensities of reflections that should be
indentical based on point group symmetry. As k,B are expected to
be smoothly varying they are often restrained during scaling. If you
turn on absorption correction it will additionally refine an empirical
absorption correction parameterized as spherical harmonics. The
scaling model is described in
Dominika Borek's CCP4 presentation from 2009. The per-frame
correction factors model things that vary primarily with image number,
of which primary absorption and scattering effects due to the
variation of the volume of the crystal in the beam are the main one,
and a B-factor term that potentially models the impact of radiation
damage on the crystal (fall-off in scattering with respect to
resolution). In reality things aren't quite that clean, since
anisotropic scattering will turn up in part in the per-frame B-factor,
and longer-term variations in e.g. detector response or beam intensity
get folded into the per-frame scale factor. An absorption correction
that is additional to this "k+B" scaling also serves to reduce errors
and is doubtless analogous to the existing methods in
XSCALE(XDS)/SCALA/AIMLESS. In any event, all scaling methods involve
exploiting the redundancy in your data as a method for estimating the
quality of the scaling result is going to improve as the redundancy of
your data improves. The estimates of the mean intensity of the
unique reflections also improves with increased redundancy.
Quality Estimates: R_merge is a simple statistic that reflects
the proportional deviation of reflections that should have the same
intensity based on symmetry. Its synonym is R_sym and they are used
interchangeably in the modern era. R_merge is systematically
under-estimated for data with low redundancy and approaches its "true"
value only in the case of high redundancy, which is inevitably a
higher R_merge. This presents the ironic situation where
low-redundancy R_merges are lower than high-redundancy R_merges for
the same data, despite the expectation that the high-redundancy data
has better-estimated intensity averages. To deal with this, R_meas
was introduced which is the redundancy-compensated version of R_merge
(see XDS
wiki) and is more reliable over a larger range of multiplicities.
See Diederichs & Karplus (1997). Scalepack now
reports R_meas as well as R_merge. Both R_meas and R_merge should be
in the low single digits for the low resolution (i.e. strongest, best
measured) data after scaling and will inevitably rise towards the high
resolution cutoff which corresponds to weaker data.
R_merge<=R_meas. The useful high resolution cutoff is a
matter of active debate but probably data can be considered useful up
to R_meas values above 60% and possibly even above 80% but up in these
"nose-bleed" ranges the statistic CC1/2 is more
useful. CC1/2 is the correlation coefficient between data
randomly split between two halves of a data set. Current
rules-of-thumb suggest that one should include data with
CC1/2 higher than 0.5, although my own experiences suggests
that I'm more comfortable with a cutoff that is around
CC1/2 ~ 0.75. Here again, however, CC1/2 is not
going to be accurately estimated if you have no redundancy in your
data. (For more on the
how-long-is-this-piece-of-string/how-far-does-my-data-go discussion
see: Evans & Murshudov (2013) and references
therein - the goalposts have been moved but you still have to use your
brain and perhaps use the paired refinement technique in order to
exploit this weaker data).
Postrefinement: Under the Global Refinement section of
the scale tab there's a variety of options that fall under the general
heading of postrefinement (so named because it occurs after the
data integration step, but "global" is perhaps a more informative
name). Recall that during indexing and during integration the
refinement of parameters - particularly unit cell dimensions - the
refinement is quite local and doesn't consider the whole of
diffraction space. Parameter refinement may quite easily fall into
false local minima during minimization. Postrefinement - activated if you
select anything above the bottom option - considers the location of
diffraction maxima on all the frames at once, so in particular it
should provide much more accurate unit cell dimensions for downstream
use. It also provides more accurate estimates of the Lorentz correction which can be large for low
resolution reflects that lie near the rotation axis. Usually you want
to do some form of postrefinement even if it's not immediately obvious
which of the options is most appropriate (and in anything except
pathological cases there is little difference).
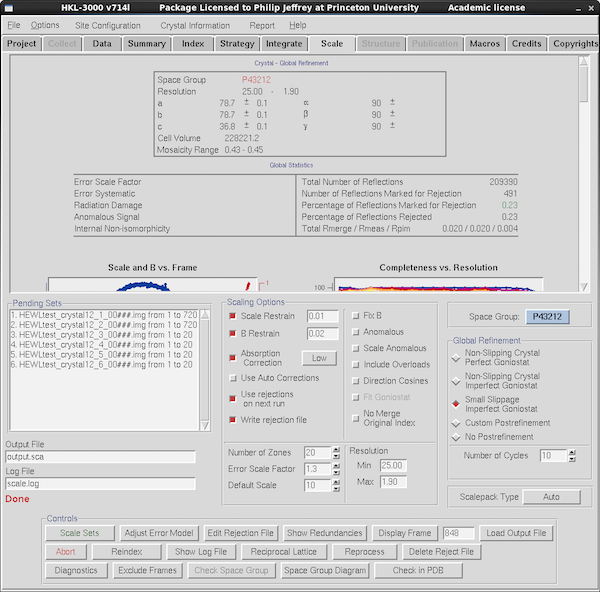
|
The actual process of scaling is easier to comprehend than the more
theoretical discussion above. Select the Scale tab. Make sure
the data runs you want to merge are included in the "Pending Sets"
list. Click on the toggles for Scale Restrain, B Restrain, Absorption
Correction (Low), Write Rejection File and over on the right Small
Slippage Imperfect Goniostat. Leave Use Rejections on Next Run
unchecked. Pick the low resolution cutoff as 25Å since the beam
stop is fairly small on this new system. If you've done any prior scaling
click the "Delete Reject File" button and then at the top left of the button
array in Controls, click Scale Sets.
What you've done is scale all the data runs lumped together,
minimizing the discrepancies between symmetry-related reflections.
It'll take a few seconds to do this and then the GUI will parse the
Scalepack log file and populate the graphs in the upper selection of
the scale tab with a summary of the Scalepack log file. You can view
the actual file (sometimes easier) using "Show Log File". But the
second table from the top - "Global Statistics" - is a pretty good
summary: 209K observations with <500 marked for rejection (the
green color for 0.23% indicates "this is good") and an R_meas of 2.0%
for data in the range 25-1.9Å. This data goes much further but
this is the acceptance criterion data set and 2.0Å is all we
need for that purpose.
By default Index (and therefore Integrate) will select the
lowest-symmetry point group consistent with the Bravais lattice you
selected in the Index step. Assuming you don't have pseudo-symmetry
or twinning this is a good choice for the initial scaling run (which
would have been done in P4 in this case) and then you can explore how
changing the space group changes the scaling statistics.
Changing the point group from 4 to 422 might change them a lot
(e.g. from space group P4 to P422) but changing between P4 to
P41 will only make a minimal difference - same point group
and P41 differs from P4 only by having some (0,0,l)
reflections systematically absent. P41
differs from P42 in the pattern of systematic absences and
P43 has the same pattern as P41 because they are
enantiomporphic space groups where the only difference is the
direction of the screw axis. So while chosing space groups be aware
that different space group choices within a point group
basically have the same scaling statistics. I knew HEWL grew in
P43212 so it was easy to select at the start but
it would be identical to P41212 and very similar
to P4322 in scaling stats - the best way to tell the actual
space group is after the scaling step when you (hopefully) solve the
structure.
Since you can solve the structure via S-SAD using this data I
should have clicked on the "Anomalous" button in the middle column so
it would output (+h,+k,+l) and (-h,-k,-l) without
merging them.
|
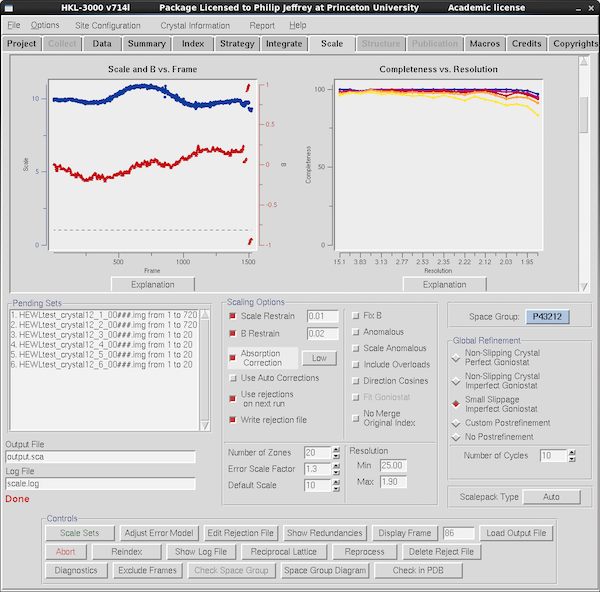
|
Scroll down in the graphical data tables to see some statistics with
resolution or by frame number. At left here is the scale factor
and B-factor by frame which you expect to move around smoothly but
perhaps jump a little at the boundaries between data runs. Here you
can see some disjoint behavior for the last few runs (20 frames each,
so 5 degrees) and you might be tempted to omit them from scaling - at
the bottom there's a button for Exclude Frames to allow you to
omit some especially troublesome frames, but in this case you'd just
go back to the Data tab and deselect the Scale checkbox for any
runs you want to delete in their entireity. At right is the
all-important completeness vs resolution table - if you want to
make any real use of your data you want it to be >85% but there's
usually little excuse for it being anything <95% unless it's in
space group P1. The color-coding is by redundancy (this data is
overkill - there's nearly always some separation) and the explanation
for the scheme is via the Explanation button, as it is for most
graphs.
|
|
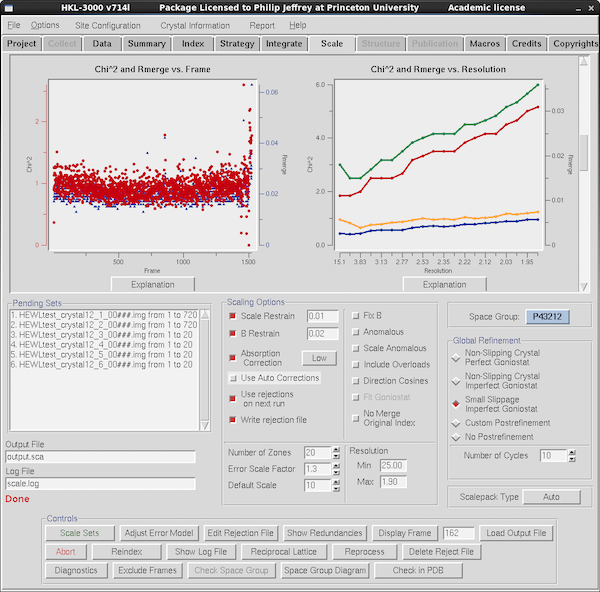
Scroll down some more and you see more data quality tables - here it's
R_merge vs resolution and Chi2 vs frame number.
Scalepack's error model expects that the Chi2 should
be 1.0 for data whose sigmas are estimated correctly. Inherently this
is comparing what you'd expect R_merge to be vs what the
observed I/σ(I) actually is, and changing the scale of the
σ(I) until the Chi2 is 1.0. You do this via the
Error Scale Factor just above the button stack - if Chi2 is
>1 increase error scale factor by 0.1 at a time until
Chi2~1. And reduce Error Scale Factor if Chi2 is
<1. You can also adjust the error scales in resolution
shells via the Adjust Error Model button. Programs like
AIMLESS and XSCALE do all this for you, so here again SCALEPACK is
needlessly manual in the adjustments. If you're doing this step you
do want to turn on Use rejections on next run to get the
best estimate of final scaling statistics, but since this excludes
outlier reflections from scaling, and the outlier criteria are based
on σ(I) you need to Delete Reject File and do 2-3 cycles
of scaling if you change Error Scale Factor significantly. You can
get obsessive about adjusting the error model so that
Chi2~1 but the most important thing is to get it relatively
close. (Ironically modern maximum likelihood refinement programs
largely ignore σ(I), although correct σ's are somewhat
useful for experimental phasing).
The other table is for R_merge vs resolution and, yes, it's using
R_merge rather than the preferred R_meas. How very retro. This data
is too good to be useful as a discussion point - the R_merge is only
3% in the high resolution bin - and normally you'd see a "ski ramp"
with a low % value on the left and your resolution limit on the right,
adjusted (the Resolution Max field) so that R_merge is in the 60% range or thereabouts.
See the discussion above for "how far does my data go". Best to click Show
Log File and look at the last table on the log file to see what
CC1/2 and R_meas are in the outermost shell and adjust the
resolution cutoff accordingly.
The final steps are: adjust your high resolution cutoff; adjust error
scale factor to make Chi2~1; check your best-guess for
space group; Delete Reject File and do 3-5 cycles of scaling to
converge on the final output; go solve your structure.
|
Downstream Data Wrangling
You can do whatever you want, but I'm a quite strong advocate of
getting out of HKL3000 and doing structure solution using
CCP4/SHELX/PHASER/PHENIX or the program of your choice. I do not
see any advantage for doing structure solution in HKL3000 and there
are quite advanced GUI and workflow for SHELX (HKL2MAP), CCP4 (CCP4I2)
and PHENIX via their respective GUIs. And HKL3000R is just using CCP4
and SHELX via its own GUIs so there's no algorithmic advantage
to using HKL3000. A while back I wrote a Data Wrangling page that would give you some
flavor of what's involved from a command-line perspective, but there
are data import workflows in CCP4i and CCP4i2, plus SHELXC deals with
Scalepack files directly. The files you want from the processing
directory are output.sca and scale.log.
HKL3000 is not an Expert System
In the previous version of
this document I waxed lyrical about the ways that HKL3000R could
have done it better - my major axes-to-grind are Profile Fitting
Radius and Spot Size and how they should be automatically adjusted,
but you can go check that out for yourself. Probably the antithesis
of the HKL3000 approach is autoProc where all I do is type "process" and some
shell script goes and makes decisions for me about data processing. I
don't necessarily agree with all of those decisions either, but it
gets me pretty close to the desired end-point without me having to
click any buttons. I've not yet tested to see if autoProc can process
this data smoothly - you may need to pull .cbf data off the
framegrabber.
Phil Jeffrey, Princeton, last modified August 18th 2017.